Rett Syndrome (RTT)
Rett syndrome is a rare, neurodevelopmental disorder that affects approximately 6,000 – 9,000 patients in the U.S.
Rett syndrome is a rare, neurodevelopmental disorder that affects approximately 6,000 – 9,000 patients in the U.S.
Rett syndrome (RTT) is a rare neurodevelopmental disorder that occurs in approximately 6,000 to 9,000 patients in the U.S.1,2 While the majority of patients are females, males who meet the criteria for RTT have also been reported.3 Individuals with RTT may require assistance for most activities of daily living.18

Individuals with RTT undergo seemingly normal development until approximately 6 months of age. Then, the manifestations of RTT begin to appear progressively over several stages: developmental stagnation (age 6-18 months), sudden developmental regression (age 1-4 years), pseudostationary/plateau (age 2 years-potentially life), and motor deterioration (age 10 years-life).13,14
They experience profound functional impairment related to the central nervous system, such as a loss of speech/communication skills (including eye contact), loss of purposeful hand use (fine motor skills), development of hand stereotypies and absent or impaired mobility (gross motor skills).13,14
RTT is a complex disorder, and individuals may experience a range of symptoms associated with the syndrome, including gastrointestinal complications, skeletal abnormalities, cardiac conduction problems, and neuroendocrine abnormalities.16,17
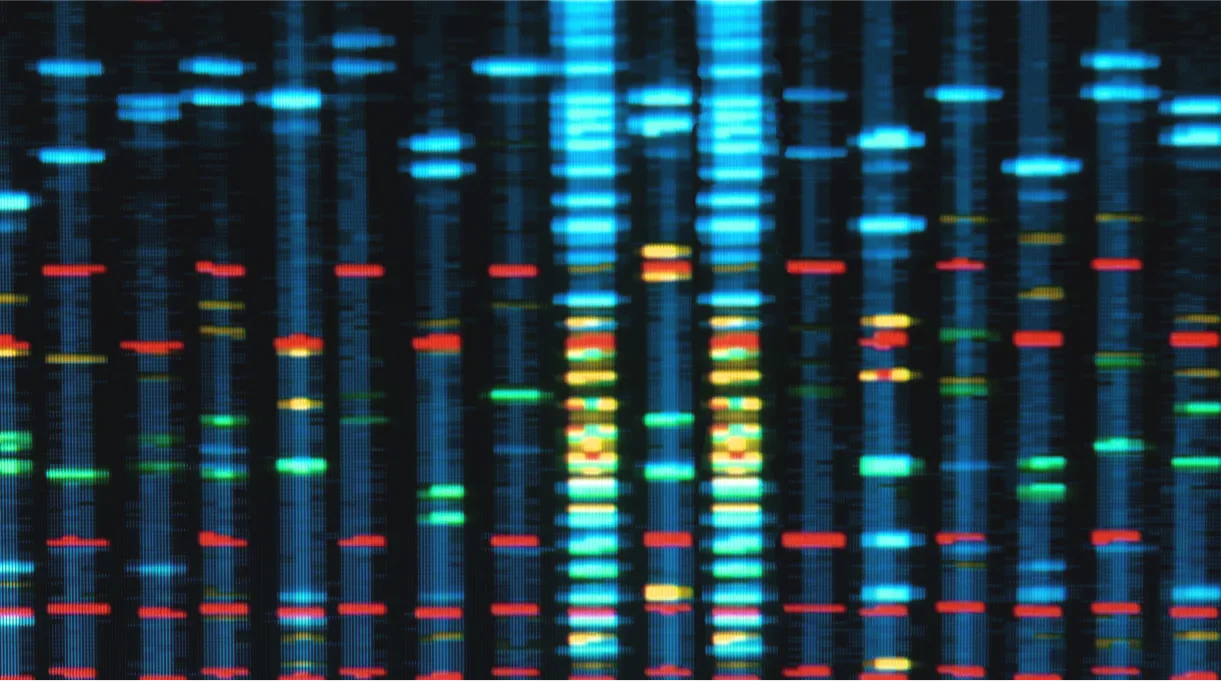
RTT is diagnosed based on clinical evaluation, typically by 3 years of age, with confirmatory genetic testing used to determine the presence and type of underlying mutations.10,11 The diagnostic criteria for RTT reflect the current understanding of the clinical features of RTT, which include disturbances in ambulation, hand use, language, growth and muscle tone abnormalities, breathing and sleep disturbances, and other manifestations.10
In ~95% of patients diagnosed with classic RTT, the disease is caused by mutations in the X-linked MECP2 gene.4 MECP2 encodes methyl-CpG binding protein 2 (MeCP2), which modulates gene expression by binding to methylated CpG dinucleotides, primarily by activating but also by repressing transcription.5-8 The activity of the MeCP2 protein is diminished in both neurons and astrocytes in individuals with RTT.9
Although underlying mutations in genes such as MECP2 are neither necessary nor sufficient for diagnosis, mutational analysis can help predict severity in order to better prepare for the needs of individuals with RTT.10,12
To report an Adverse Event or Product Complaint please call (844) 422-2342.